Necrosis
Author(s): Tim Kendall
Learning outcomes
By the end of this CAL you will be able to:
- Understand the stimuli that lead to death by necrosis
- Recognise the morphological features shared by necrotic cells
- Describe the mechanisms and sequence of events in necrosis
- Describe specific types of necrosis and their clinical context
Introduction Part 1 of 10
Necrosis is the pattern of cell death that occurs in response to injuries such as hypoxia, extremes of temperature, toxins, physical trauma, and infection with lytic viruses.
The injury to a cell is said to be irreversible if it kills the cell. If the damage is a bit less, the injury is said to be reversible. In cell injury, there’s a continuum of sublethal changes, which may progress to lethal cellular changes, depending on the severity and duration of the injurious stimulus.
Cell injuries Part 2 of 10
Necrosis is the pattern of cell death that occurs under violent circumstances. These include:
- hypoxia
- extremes of temperature
- toxins at high doses
- complement
- physical trauma
- infection with lytic viruses
All of these cause severe disturbance of the cellular environment leading to death.
Reversible cell injury Part 3 of 10
The initial cellular response to injury is rapid, leading to cell swelling. If the injury is minor and short-lived, the cell swelling can reverse and the cell return to normal; this early swelling is an example of reversible cell injury.
Another reversible cell injury is steatosis in hepatocytes of the liver.
The normal liver contains hepatocytes arranged in plates. The hepatocytes are neither swollen nor steatotic (fatty).

In fatty liver disease, from either alcohol or non-alcohol related injuries such as diabetes, high lipids, or obesity, both cell swelling and steatotic reversible injuries are evident.

If the injury ceases, these changes can rapidly resolve.
Infarction - an example of necrosis Part 4 of 10
If the injury is too severe or prolonged, death by necrosis occurs. Cell death by necrosis is central to many pathologies, including most of the common diseases.
If there is reduction or cessation of the blood supply to a tissue (ischaemia), then a zone of that tissue dies by necrosis, and the process is termed infarction. A particularly important site where this occurs is the heart and a myocardial infarction results.

Cut surface of a slice through the left ventricle of the heart. Following occlusion of a coronary artery, a zone of myocardium becomes severely hypoxic and the cells die. The result is seen here as a pale area (arrow) within the wall of the left ventricle. Sheets of cells in continuity die together and the overall tissue structure becomes disrupted.
Cellular morphology Part 5 of 10
Cytoplasmic changes
Following sublethal, early reversible injury, the cell swells up. Early changes include blebbing of the plasma membrane, dilatation of the endoplasmic reticulum with loss of ribosomes, cytoplasmic swelling (intracellular oedema).


With a continued injury, such as a reduced oxygen supply (hypoxia), the changes become irreversible. These changes include rupture of the plasma membrane, mitochondrial swelling, calcium matrix density formation in mitochondria, lysosomal breakdown producing autodigestion, and cellular degradation.

Let’s recap some key points of the sequence of events. As the cell reaches the “point of no return”, necrosis becomes inevitable. This coincides with two changes in the mitochondria: high amplitude mitochondrial swelling, and the formation of calcium matrix density.

The necrotic cell swells rapidly and ruptures. Both the plasma membrane around the outside of the cell and the internal membranes round the organelles can burst. Cytoplasmic organelles spill out and can initiate inflammation. Neutrophil polymorphs are attracted to the site.
Nuclear changes
Nuclear changes in necrotic cells include some coarsening of the heterochromatin (dark chromatin). But the usual distinction between heterochromatin and euchromatin (light chromatin) is retained. Eventually, the chromatin degrades (karyolysis) and the nucleus appears as an outline of its former self.
Electron microscopic appearances
We can readily recognise some of these changes by electron microscopy. Before we look at a necrotic cell, here are two normal cells. Note the usual nuclear structures of heterochromatin (darkly staining) (a), and euchromatin (lightly staining) (b).

By contrast, here’s a necrotic cell, it has the following: dilated endoplasmic reticulum (a), coarsened, darkly staining heterochromatin (b) within the nucleus, but still easily distinguished from the lighter staining euchromatin (c), “Washed-out” appearance of the cytoplasm (d).

Light microscopic appearances
We can also recognise necrotic cells by light microscopy. Let’s look at an example in a histological section of an infarct in the heart: a myocardial infarct. Histology of this myocardial infarct shows loss of cytoplasmic detail (loss of striations in some of the cells) with the formation of an eosinophilic (a) “ground glass” appearance. As chromatin degrades, nuclear staining fades (karyolysis) (b) forming ghost nuclei.

Molecular events and consequences Part 6 of 10
There are three central mechanisms leading to cell rupture in necrosis:
- loss of selective plasma membrane permeability.
- loss of calcium homeostasis.
- failure of membrane ion pumps.
These three are interlinked. Any one event often leads on to another, allowing fluid and ions into the cell. This process spirals out of control: a vicious circle occurs and the cell swells up until it finally ruptures.

Loss of membrane integrity
Calcium ion flux
As membrane function is lost, necrotic cells accumulate calcium ions. Calcium ion flux leads to calcium matrix density formation in the mitochondria.
Calcium ions also activate enzymes:
- proteases, which can break down membrane protein pumps.
- Phospholipases, which can attack lipids in the plasma membrane.
This leads to a vicious circle, producing progressive loss of membrane integrity and cell rupture.
Role of ATP
There is an important relationship between ATP levels and membrane permeability. Injured cells often become ATP depleted, such as in hypoxia with lack of oxygen for the respiratory chain enzymes to generate ATP.
Here, ATP content versus cell permeability is shown, for ischaemic cardiac muscle.

This loss of ionic gradient maintenance allows the osmotic influx of water into the cell, initially as reversible hydropic change. If unchecked, this eventually produces membrane damage and increased cell permeability, and, finally, rupture.
Other causes of increased cell permeability include injury to the cell membrane from:
- Physical trauma.
- Freezing or boiling.
- Complement.
- Lytic viruses.
Reactive oxygen intermediates
As well as hypoxia producing ATP depletion, it can also induce the formation of Reactive Oxygen Intermediates (ROI). Hypoxia, ionising radiation and xenobiotics (some drugs, etc) can generate ROI.
Reactive Oxygen Intermediates (ROI) are highly reactive free radicals which can attack many biological molecules.
ROI generate DNA strand breaks. This induces the enzyme poly ADP ribose polymerase (PARP). This enzyme reacts with broken DNA ends, adding ADP-ribose to the broken ends. This further depletes the cell of ADP/ATP.
ROI free radicals attack proteins (via their thiol -SH groups) such as membrane pumps, thus damaging membrane permeability. ROI free radicals also attack membrane lipids (by peroxidation), thus further damaging membrane permeability.
Mechanism of necrosis - summary Part 7 of 10
By putting all of these elements together, we can see the interlinking of the 3 major mechanisms:
- membrane permeabilisation
- ATP depletion
- ROI generation
These changes feed into the vicious circle of:
- membrane permeabilisation
- calcium ion influx
- defective membrane pumps
This leads to swelling and rupture of the necrotic cell.
ROI – summary
To summarise, then: the important Reactive Oxygen Intermediates (ROI) in cell injury are:
- superoxide O2–
- hydrogen peroxide H2O2
- hydroxyl radical OH
Normally, cellular protective mechanisms limit the damage caused by ROI. The agents that inactivate them are:
- anti-oxidant tocopherol (vitamin E component)
- catalase ( H2O2 to H2O & O2)
- superoxide dismutase (2O2– & 2H+ to H2O2 & O2)
- glutathione peroxidase (glutathione, GSH, soaks up ROI becoming GS-SG)
Thus, oxidative stresses can injure cells by blocking these protective mechanisms, allowing unlimited damage by ROI.
Reperfusion Part 8 of 10
In certain circumstances, the blood supply to a tissue is cut off and then restored. This leads to reperfusion of ischaemic tissue, which may sometimes cause further tissue damage (Ischaemia-reperfusion injury).
Reperfusion of ischaemic organs can induce further necrosis from a burst of ROI. Xanthine, derived from metabolised ATP, accumulates in ischaemic cells and there is proteolytic cleavage of xanthine dehydrogenase to xanthine oxidase in the endothelial cells lining blood vessels.
Fresh oxygen, brought by the reperfusion, allows xanthine to be oxidised by xanthine oxidase, generating ROI, causing endothelial damage, oedema and further necrosis.
Inflammation occurs if cell contents spill out after the loss of membrane integrity.
Morphological patterns of necrosis Part 9 of 10
Classical morphological patterns of necrosis can be defined:
- Coagulative
- Liquefactive
- Caseous
- Gangrenous (dry)
- Fat
These patterns are only visible as a consequence of changes that occur to the necrotic tissue in a living host.
Coagulative necrosis
After necrotic cell, proteins can denature (coagulate) to leave ‘ghost’ outlines behind. The dead cells lose their nuclei and may stain more intensely.
This pattern of coagulative necrosis is seen in many tissues, for example in the myocardium after infarction (necrosis following ischemia).

Liquefactive necrosis
Infiltration of dead tissue by large numbers of neutrophils leads to digestion (rather than coagulation) of cell proteins. This leads to loss of normal tissue architecture and is known as liquefactive necrosis.
This can occur when there is a secondary bacterial infection of the dead tissue when it is called wet gangrene. Liquefactive necrosis is common after cell death in lipid-rich tissue such as the brain (cerebral infarction).

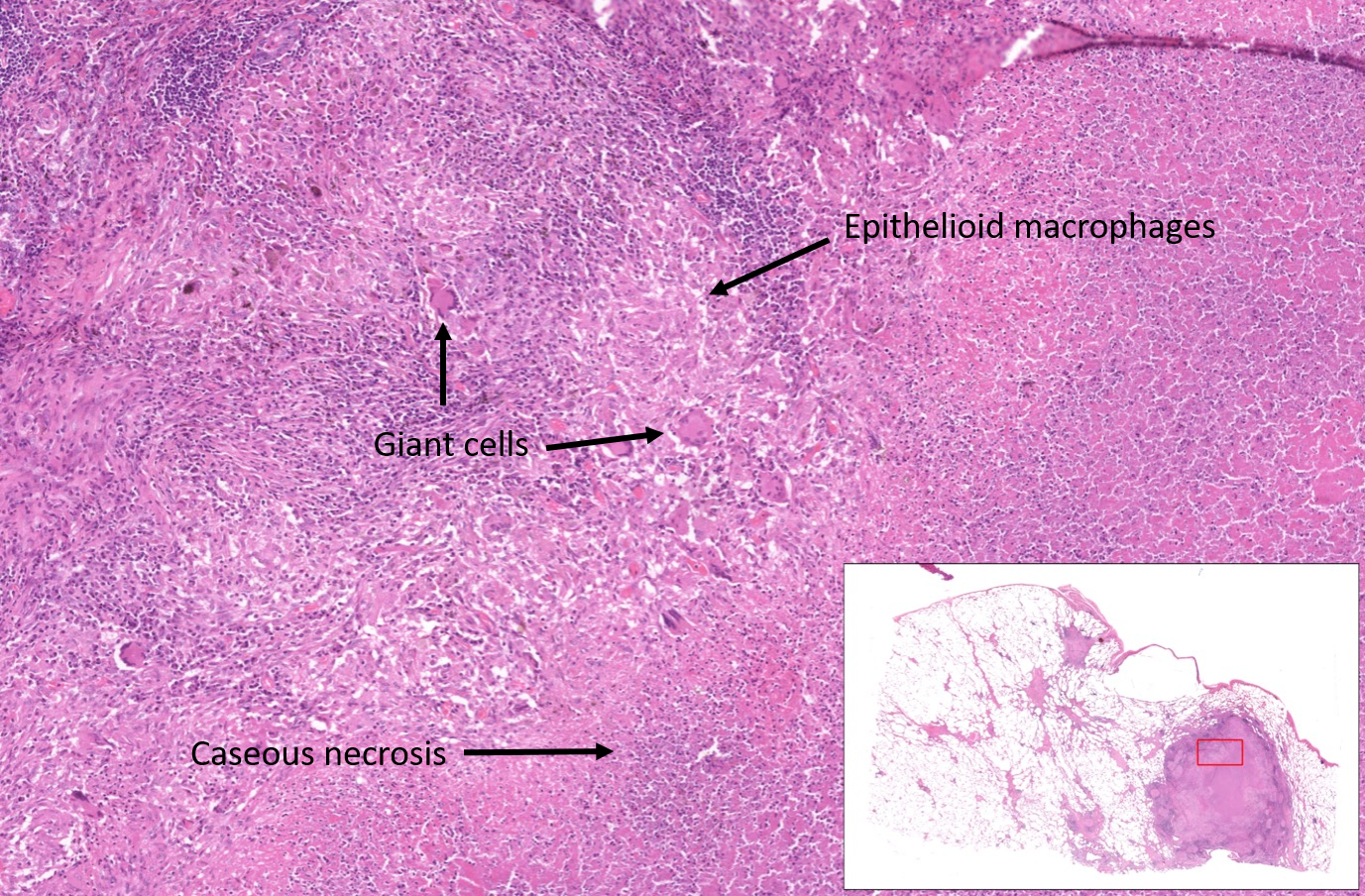
Caseous necrosis
Granulomas are aggregates of epithelioid macrophages and giant cell macrophages, often surrounded by lymphocytes.
Granulomas are found as a response to foreign bodies, in some autoimmune diseases, and in mycobacterial infection (e.g. M.tuberculosis).
Necrosis can occur in the centre of granulomas, typically in mycobacterial infection. This is described as caseous necrosis because the macroscopic appearance was considered to be cheese-like.
Dry gangrene
Dry gangrene is coagulative necrosis of an extremity due to slowly developing vascular occlusion. This can be seen in patients with diabetic vascular disease.

Fat necrosis
Fat necrosis is a pattern of necrosis that occurs due to degradation of fatty tissue by lipases (released from dead cells) to form chalky deposits.
This can be seen in acute pancreatitis (acute inflammation of the pancreas causing necrosis of pancreatic acinar cells and lipase release), or from trauma to fatty tissues.
