Acute Inflammation and Innate Immunity
Author(s): David Dorward and Heather Lockwood
Learning outcomes
Following the completion of this tutorial, you should be able to:
- Define innate immunity and appreciate its role in the body’s immune response and acute inflammation
- Describe the components of the innate immune system and provide an overview of their function
- Describe the early and late events of the innate immune response and understand how each is initiated
- Appreciate how innate immune cells stimulate acute inflammation and pathogen killing
- Understand, and be able to discuss, the pathophysiology of acute inflammation and how this manifests clinically
- Discuss how disorders of innate immunity may contribute to disease
What is innate immunity and why do we need it? Part 1 of 7
The principal function of the immune system is to identify and eliminate microorganisms and other harmful substances and/or cells.
The immune response to infection may be divided into three phases:
- The innate phase
- Early induced innate response
- Adaptive immune response

The innate immune system is the body’s first line of defence against invading pathogens and may be thought of as the first ‘arm’ of the immune system as it responds quickly (within minutes – hours) to pathogenic assault in a relatively non-specific manner.
The innate immune system comprises a range of tissues, cells and soluble factors that, along with the adaptive immune system, work together to identify, kill and eliminate disease-causing pathogens (e.g. viruses, bacteria, fungi and parasites) and cancer cells. It is essential that innate immune cells can recognise and respond to pathogens and are able to distinguish ‘self’ from ‘non-self’ molecules. This ‘recognition phase’ is achieved via the expression of Pattern-recognition receptors (PRRs) on innate cells which recognise conserved Pathogen-associated molecular patterns (PAMPS) expressed by pathogens.
Pattern recognition receptors include the Toll-like receptors, nucleotide oligomerisation receptors (NLR), C-type lectin domain family members and the RIG-1 like receptors (RLR). Pathogen-associated molecular patterns include viral and bacterial nucleic acids, pathogen cell wall components and flagellar proteins.
| Pattern – recognition receptors (PRRs) | Pathogen-associated molecular patterns (PAMPs) |
| Toll-like receptors (TLR) | Bacterial lipids, viral RNA, bacterial DNA, bacterial and parasite proteins |
| Nucleotide oligomerisation receptors | Bacterial and viral RNA, uric acid, β amylase |
| C-type lectin receptors | Muramyl dipeptide (Mycobacterium tuberculosis) |
| RIG-1 like receptors | Viral RNA |
| Common Pattern recognition receptors and examples of corresponding pathogen-associated molecular patterns | |
In addition to PAMPs, the innate immune response may also be initiated by cell-derived, endogenous molecules called Damage-associated Molecular Patterns (DAMPs) which are released in response to tissue injury and/or cell stress. The recognition of DAMPs and PAMPS by innate cells results in the activation of effector cells, acute inflammation and removal of the infectious agent.
Even before the early induced innate response, the innate immune system is able to immediately respond to new pathogenic infection by the release of preformed soluble molecules already present in bodily fluids (e.g. extracellular fluid, epithelial secretions, blood). These molecules include antimicrobial enzymes (e.g. lysozyme) and peptides (e.g. defensins) which attack, lyse and digest bacterial cell walls to either weaken or kill the infecting pathogen.
The innate immune and early induced innate responses have no capacity for immunological memory and as such confer no long-lasting protection against infection. In contrast, immunological memory is a fundamental feature of the adaptive immune response but this can only be induced once the innate immune defences are breached.
Barriers of the innate immune system Part 2 of 7
Anatomical barriers
Anatomical barriers consist of physical, chemical and biological agents that block entry of environmental pathogens before they can infect the host. For a pathogen to invade the body, it must first bind to or cross an epithelial barrier. However, the epithelial surfaces of the body (e.g. skin, gut mucosa) are extremely effective barriers to infection as they are highly specialised to carry out this protective function.
Epithelial anatomical barriers and their specialised protective functions
Skin
Stratified squamous epithelium with surface keratin which creates a dry, hostile environment for microbial growth and resists pathogen invasion via regular desquamation. Epithelial cells produce fatty acids, lamellar bodies and β – defensins which inhibit microbial growth and proliferation.

Gut mucosa
Epithelial cells joined by tight junctions. Unidirectional flow of gut contents (fluid) to impede microbial adhesion and facilitate removal of pathogens. Low pH, enzymes (e.g. pepsin), mucin and soluble factors (e.g. defensins) act as chemical factors to impede microbial growth/proliferation. Goblet cells secrete mucin which traps microbes and helps to facilitate their removal from the body.

Respiratory mucosa
The mucociliary action traps pathogens and extrudes them from the body. Type II pneumocytes produce pulmonary surfactant to further prevent and inhibit microbial growth/proliferation.

Eyes/nose/oral cavity
Tears, saliva, nasal cilia provide mechanical protection by preventing microbial attachment/facilitating their removal. Tears and saliva also contain enzymes (lysozyme) and soluble factors (e.g. defensins) that assist in microbial cell wall lysis and pathogen killing.

In addition to the features above, commensal microorganisms also function as a protective barrier as they compete with pathogenic microorganisms for resources and some produce antimicrobial agents (e.g. acids) to inhibit or kill rival pathogens.
In health, most epithelial surfaces have a population of commensal bacteria and their presence helps to strengthen epithelial defences by stimulating epithelial cells to produce their own antimicrobial peptides (e.g. lysozyme).
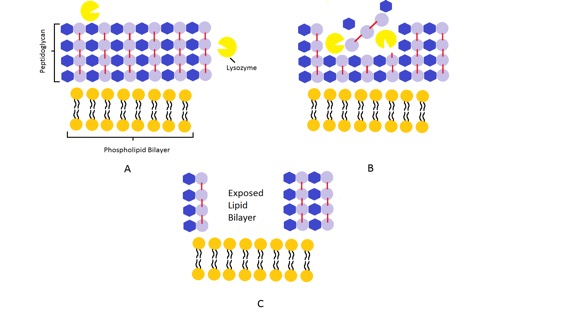
Neutrophils Part 3 of 7

Neutrophils are attracted to the site of infection via a range of chemical mediators (chemoattractants) which direct neutrophil migration in a process called chemotaxis.
Common chemoattractants include complement and cytokines. Neutrophils are a type of polymorphonuclear leukocyte (white blood cell) which are one of the first innate cell types to be recruited to the site of infection. Once at the affected tissue, they perform several important functions including ingestion (phagocytosis) and killing of pathogens, removal of necrotic tissue and recruitment of other innate immune cells.
Neutrophil recruitment
In health, neutrophils reside in the blood and travel within blood vessels close to the vessel wall. When a tissue becomes infected by a pathogen, resident sentinel leukocytes release a range of inflammatory mediators which induce endothelial surface changes and initiate neutrophil recruitment. In addition, endothelial cells may also be directly activated via PRR-mediated pathogen detection which increases adhesion molecule expression within minutes.

The sequence of events in neutrophil recruitment from the vessel lumen to infected tissue may be divided into the following steps:
- Margination, tethering, rolling and endothelial adhesion.
- Endothelial transmigration
- Migration to affected interstitial tissue
Margination
In the presence of acute inflammation, blood flow slows (stasis) and vessel wall shear stress falls leading to an accumulation of neutrophils and other leukocytes at the endothelial surface (margination). The endothelial surface also changes under the influence of inflammatory mediators (e.g. histamine, cytokines) to facilitate leukocyte adhesion via the up-regulation of adhesion molecules (e.g. P-selectin, E-selectin) which bind to selectin ligands expressed by neutrophils (e.g. P-selectin glycoprotein ligand 1 (PSGL1)).
Tethering and Rolling
The up-regulation of endothelial adhesion molecules results in the transient tethering (capturing) of free-flowing neutrophils to the vessel wall which subsequently ‘roll’ along the endothelium in the direction of blood flow. Neutrophil rolling is facilitated by the rapid and balanced formation and dissociation of adhesive bonds at the front and rear of the cell respectively (Figure 4). As neutrophils roll along the endothelium, they are exposed to a range of chemokines (e.g. the ELR-CXC chemokines CXCL8 (IL-8), CXCL1 (KC), CXCL2 and CXCL5 (LIX)) which initiate neutrophil activation and promote firm endothelial adhesion.
Firm endothelial adhesion
Chemokines are themselves anchored to the endothelium via their binding to heparin sulphate proteoglycans which results in an intravascular chemotactic gradient to direct neutrophil migration. This also increases the affinity of neutrophil-bound integrins (e.g. LFA1, VLA-4) for their ligands which include endothelial cell adhesion molecules (CAMs) (e.g. intercellular adhesion molecule 1 (ICAM1), vascular cell adhesion protein 1 (VCAM-1)). The binding of LFA1 to ICAM1 is essential for firm adhesion of neutrophils to the endothelium.
Transmigration
Once firmly adhered, some neutrophils demonstrate ‘probing’ behaviour whereby they elongate and insert pseudopods to actively scan their surroundings and find an optimum site for transmigration (diapedesis). Endothelial cells in the proximity of transmigrating neutrophils also undergo cytoskeletal changes to become less adherent to the extracellular matrix and thus facilitate neutrophil passage.
The transmigration of neutrophils from the vasculature to tissues is usually paracellular (i.e. between endothelial cells at cell-cell junctions); however they may also cross through endothelial cells (i.e. transcellular) (Figure 4) but the latter mechanism is less efficient and not preferred by neutrophils. Once through the endothelium (which takes about 2-5 minutes) neutrophils must then cross the basement membrane to enter the extravascular space which takes approximately 5-15 minutes. The process of transmigration is mediated by the complex interaction between CAMs, integrins, junctional proteins and other endothelial cell molecules.
Migration – chemotaxis
Once in the interstitial tissue, neutrophils must then travel to the site of tissue injury. This is achieved by the process of chemotaxis in which chemical gradients attract neutrophils and direct their migration. Chemical gradients may be formed from a range of exogenous and endogenous chemoattractant substances. Common exogenous chemoattractants include bacterial products (e.g. peptides, lipids) which are released in the immediate vicinity of infecting bacteria. Endogenous chemoattractants include complement system components (e.g. c5a), cytokines, chemokines and leukotrienes (e.g. leukotriene B4).
All of the chemoattractant agents mentioned bind to G-protein coupled receptors (GPCRs) on the surface of neutrophils (and other leukocytes) with subsequent activation of a range of hierarchal signalling pathways (e.g. phosphatidylinositol 3-kinase (PIK3), p38 mitogen-activated protein kinase (MAPK)) which promote directional leukocyte migration and recruitment to the site of injury.
The above stages of leukocyte recruitment to injured tissue are essentially the same for monocytes (which become macrophages in tissues), eosinophils, basophils and lymphocytes.
Phagocytosis and pathogen killing
Once at the site of tissue injury, neutrophils (and other phagocytic leukocytes e.g. macrophages) must identify, attack and ultimately kill the offending pathogen.
Neutrophils – clinical context
Clinically, the presence of high numbers of neutrophils leads to pus formation (suppurative inflammation) which is usually indicative of bacterial infection.
Therefore, if you are interpreting a full blood count (FBC), a high white cell count with high numbers of neutrophils (neutrophilia) should make you suspicious of bacterial infection. However, it is important to remember that neutrophil count may also be raised in non-infectious conditions (e.g. burns) or metabolic disorders (e.g. diabetic ketoacidosis) so it is essential that the FBC results are interpreted along with available clinical information.
Complement Part 4 of 7
The complement system is a collection of approximately 30 plasma proteins that function together to attack pathogens both directly and via promotion of inflammation.
Complement components (numbered C1 through C9) are produced by the liver and circulate in the plasma in an inactivated form before entering infected/injured tissues where they are enzymatically cleaved and activate other downstream complement proteins as part of the complement cascade.
Of all the complement components, C3 and C5 are the most important mediators of acute inflammation and can be activated by multiple proteolytic enzymes present in inflammatory exudates in addition to the mechanisms discussed above.
| Complement component | Principal activity |
| C5a | Neutrophil activation and chemotaxis |
| C3a, C4a, C5a | Increased vascular permeability, degranulation of mast cells and basophils |
| C3b, C4b | Opsonisation of pathogens and immune complexes |
| C5b | Bacterial and foreign cell lysis |
| Principal activities of common complement components | |
A critical step in initiating the complement cascade is the cleavage of C3 into two functionally distinct fragments, C3a and C3b.
This occurs via one of three pathways:
- The classical pathway
- The alternative pathway
- The MB-Lectin pathway
Although these pathways differ in their mode of initiation they converge to activate the same effector molecules and thus have the same end result.
Mechanisms of action
The complement system protects against infection in three main ways. Firstly, large numbers of activated complement proteins (in particular C3b and C4b) are generated which covalently bind to pathogens in the process of opsonisation. This enables neutrophils and macrophages to recognise and bind to the complement and subsequently engulf (phagocytose) the pathogen.
Secondly, certain complement fragments (especially C5a) act as chemoattractants to recruit phagocytes to the site of complement activation and augment the acute inflammatory reaction.
Finally, late complement components (e.g. C5b, C6-C9) are able to lyse bacteria and other foreign cells via the formation of a membrane attack complex.
The activation of complement is highly regulated by several proteins (e.g. C1 inhibitor, decay accelerating factor, CD59) to prevent complement-related damage to healthy tissues adjacent to the site of injury. These regulatory proteins are expressed on normal host cells and inhibit the production of activated complement components and/or remove fragments that bind to normal cells.
Complement – clinical context
In a patient with acute suppurative meningitis, a bacterial infection of the meninges leads to activation of the alternative pathway with cleavage of C3b from C3 with subsequent activation of C5 which is then cleaved into C5a and C5b. C5a is a powerful chemoattractant which attracts neutrophils to the site of infection (in this case the meninges) and initiates an acute inflammatory response.
After approximately 7-8 days the adaptive immune system is activated and produces antibodies against the bacteria which bind to its surface and activate the classical pathway via the binding of complement. This again results in the release of C5a which augments the inflammatory reaction.
Cytokines and chemokines Part 5 of 7
Cytokines and chemokines are a collection of proteins and peptides produced in response to inflammation, infection or tissue injury which play an integral role in leukocyte recruitment and regulation of the inflammatory process. Cytokines may be classified according to the nature of the immune response (Table 4) with key pro-inflammatory cytokines including interferons, interleukins and Tumor Necrosis Factors (TNF).
Adaptive immunity: IL-2, IL-4, IL-7, IL-19, IL-15, IL-21
Pro-inflammatory signalling: IL-1, IL-6, TNFα, IL-17, Type I, II and III IFN
Anti-inflammatory signalling: IL-10, IL-12
Vascular changes in acute inflammation Part 6 of 7
Vascular dilatation
In acute inflammatory states, vascular dilatation occurs as a consequence of vasoactive mediator (e.g. histamine, serotonin) and nitric oxide release from innate and endothelial cells respectively. Vasodilatation gives rise to the redness/erythema (rubor) and increased heat (calor) seen in acute inflammation.
Increased vascular permeability
Vascular permeability is also increased resulting in the formation of acute inflammatory exudate which manifests clinically as tissue swelling/oedema (tumour). As a consequence of increased vascular permeability, the blood vessels essentially become more ‘leaky’ which allows large plasma proteins and fluid to leak from the intravascular space to the extracellular place with resultant oedema formation.
Summary Part 7 of 7
- The cardinal features of acute inflammation are calor (heat), dolor (pain), tumour (swelling), rubor (redness) and functio laesa (loss of function)
- Increased vascular permeability is a hallmark of acute inflammation